How atomistic simulation can help find a new material design solution in a few hours to days
Bosch Research Blog | Post by Mordechai Kornbluth, Soo Kim, Charles Tuffile, 2021-03-04


The latest advances in revolutionary technology are closely linked to and driven by new materials design – but how do we explore them all?
The trial-and-error method of material design via experiments is usually slow, expensive, and difficult to optimize. Hence, the idea of using high-throughput computational materials screening and machine learning is increasingly regarded as a very attractive alternative. In computational materials science (CMS), atomistic-based simulations have been recognized as a very useful tool and a complementary effort to performing experiments. In the past, identifying materials with a certain desired property via a CMS approach had not proved efficient, mostly due to the high computational costs of running these simulations, which can take months to years. Thanks to increased computational speed and power, we can now perform such computational material designs within time frames of days to weeks. When combined with publicly available, large material quantum databases, it is now possible to derive the relationships between structure, processing, properties, and performance of newly-designed materials for targeted device applications within seconds or hours.
Month- to yearlong simulations now run within days to weeks thanks to the increased computational speed and power
First-principles density functional theory calculations
As an example, tremendous research efforts are put into the search for energy-efficient and cost-competitive water desalination technologies. A large volume of salt water is processed to yield freshwater in a desalination device, where the most commonly used salts include sodium chloride (NaCl). A handful of new electrode materials that can collect cations (such as Na+, Ca2+, and/or Mg2+) have been experimentally synthesized in the last ten years. In contrast, host electrodes that can collect anions such as Cl- have remained mostly unexplored. Using first-principles density functional theory (DFT) calculations, we have identified a number of promising electrode materials configured to reversibly store and release target anions such as Cl- from saline water.
We also tested their chemical stability against decomposition within the stability window of water to avoid water electrolysis. An alternative method in desalination technology is to use an anion exchange membrane (AEM) that rejects cation transport through the AEM while allowing the anion transport. Using atomistic DFT simulation, we were able to estimate the cation (e.g., Na+, K+, Mg2+, Ca2+) and anion (Cl-) adsorption voltages in graphene oxide (GO) material. We found that while cations lead to strong binding on the oxygen functional group of GO sheets, anions such as Cl- are not bound by the GO, enabling fast anion diffusion though the GO-based AEM. Lastly, we assessed with atomistic DFT calculations that expanded graphite – with a plurality of graphene layers with an interspacing greater than 0.34 nm – can accommodate 20 to 30 times more capacity than a pure graphite, making it more suitable as a desalination electrode material.
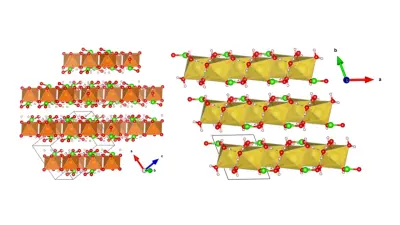
Figure 1. Illustration (left) of a CMS-designed crystal structures of anion (Cl-) intercalation host materials that can be used in desalination cleaning devices.
Figure 2. Illustration (right) of a CMS-designed graphene oxide (GO)-based anion exchange membrane (AEM), enabling selective ion (de-) adsorption chemistries.
In fact, computational compilation of materials properties such as voltage, adsorption, diffusivity, and chemical stability via atomistic DFT bulk calculations has helped understand the underlying mechanism for a number of different product applications. While exploring all new possibilities by substituting chemical elements from the periodic table in order to find materials with better structural stability, coherent mixing, or less tendency for decomposition are quite exciting, many critical problems cannot be solely addressed by DFT calculations at bulk scale.
As an example, we have found via experiments that doping the oxidized surface portion with a liquid containing a fluorine-containing salt makes the fluorinated metal surface more hydrophobic. We were able to identify that the presence of fluorine on surface metal oxides (e.g., Cr2O3 and NiO) can increase hydrophobicity by constructing an oxide and water interface using complex DFT slab models. Another example can be found in designing an anticorrosive and conductive oxide material. Generally speaking, electrically insulating material is found to be chemically inert. However, we were able to computationally predict a new ternary oxide composition that is electrically conductive and anticorrosive at the same time, followed by experimental verification.
Atomistic methods and “Big Data” in quantum databases
Lastly, I would like to comment on utilizing both atomistic methods and “Big Data” in quantum databases to address more difficult technical challenges. This new approach involves analyzing several hundred thousand existing and prototype structures, but without significant CPU time. This combinatorial approach can help to a new material solution within a few hours to days. In Bosch Research’s Computational Materials Science and Engineering group, we have addressed a number of different materials challenges including but not limited to: corrosion, coatings, oxidation, chemical reactions, electrochemical reactions, alloys, heat treatment, and decomposition.
Utilizing atomistic methods and “Big Data” in quantum databases can help to find a new material solution within a few hours to days by analyzing several hundred thousand existing and prototype structures without significant CPU time
Outlook
Overall, our atomistic simulation can help provide mechanistic understanding and reduce the number of experiments and save resources in the near future. With emerging technology such as next-generation AI, GPU, TPU, and quantum computing, our CMS approach will further create a landscape of opportunities for current and future Bosch applications that are “Invented for Life”.
What are your thoughts on this topic?
Please feel free to share them via LinkedIn or to contact us directly.
Author: Mordechai Kornbluth
Mordechai is a Senior Research Engineer in the Computational Materials Science and Engineering group at Bosch Research in Cambridge, MA. He specializes in modeling materials down to the constituent atoms and electrons in order to design and improve nanoscale properties. From sensors to semiconductors, Mordechai’s work focuses on the intersection between chemistry, physics, and engineering.

Author: Charles Tuffile
Charles is Senior Manager of the Computational Materials Science and Engineering (CMSE) group in the Research and Technology Center North America. He leads the CMSE group, which researches, identifies and develops next generation materials to deliver performance in future and current Bosch products such as fuel cells, semiconductors, MEMS devices, and DNA sequencing.

Author: Soo Kim
Soo worked at Bosch Research until June 9, 2021. Prior to joining the Energy Technologies Department in the Bosch Research and Technology Center North America, Soo worked on a number of different materials synthesis and simulation methods in the industry, in a national laboratory and in academia. At Bosch, he was mainly responsible for analyzing technical challenges experienced by other researchers or business units, and then coming up with (an) innovative material solution(s). In 2018, 2019, and 2020, he had the highest number of U.S patent filings to show for in the Energy Technologies Department.



